
Editor's Note
Slide 1 of 10
Though published in 2005, the New England Journal article by Krause and Van Etten on mechanisms of action of tyrosine kinase inhibitors (TKIs) remains a classic in oncology education, particularly the extraordinary graphic depictions of the NEJM artists. We reproduce these figures along with another graphic in an article by Rini et al on VEGF TKIs in another attempt to assist busy clinicians attempting to understand the rapidly evolving translational database that is permeating contemporary oncology.

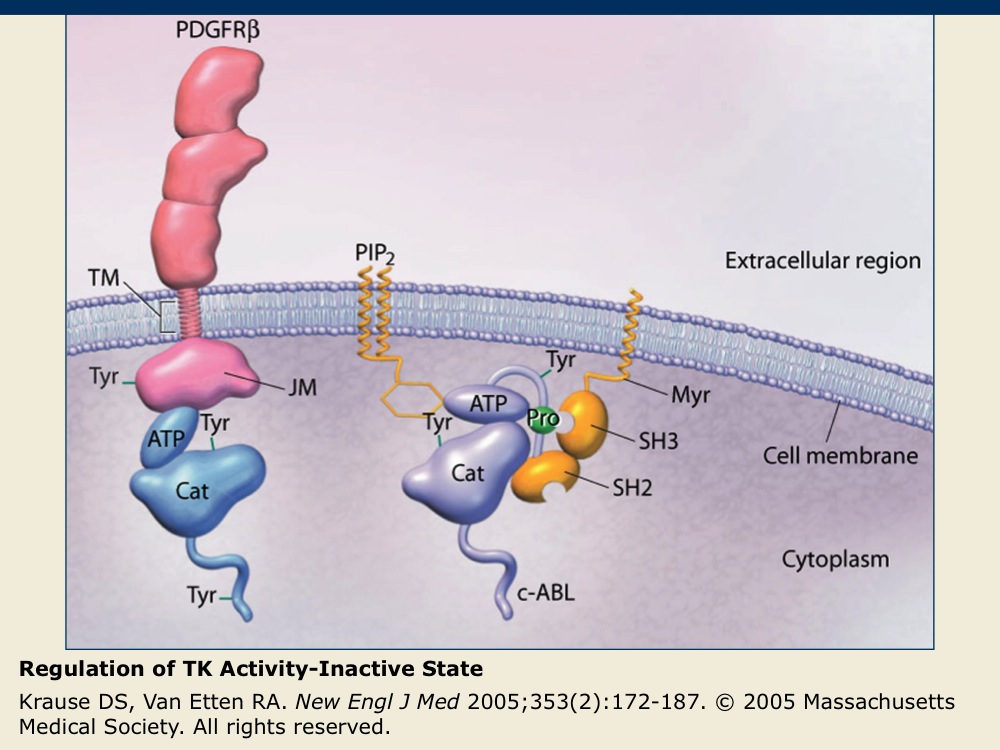
Regulation of TK Activity-Inactive State
Slide 3 of 10
The inactive PDGFR-β receptor TK exists as a single membrane-spanning molecule in which the regulatory tyrosine (Tyr) amino acids are not phosphorylated.
The inactive nonreceptor TK c-ABL is tethered to the cell membrane by PIP2 that acts as an inhibitory molecule. Nonreceptor TKs are maintained in their inactive states through interactions with inhibitory lipids or cellular proteins.

Regulation of TK Activity-Activated State
Slide 4 of 10
After PDGFR-β binds its ligand, dimerization occurs, allowing phosphorylation of the regulatory tyrosine residues. The phosphorylated tyrosines are binding sites for other cell signaling proteins, such as c-SRC and phospholipase C-γ (PLC-γ), that function in initiating downstream signaling cascades. These signaling cascades regulate cellular processes, such as angiogenesis, cell proliferation and cell survival.
For nonreceptor TKs such as c-ABL, activation may occur in response to various intracellular signals that can lead to the dissociation of the inhibitory molecules and the phosphorylation of regulatory tyrosines. Similar to the receptor TKs, phosphorylation of the regulatory tyrosines also leads to the initiation of cellular signaling cascades affecting cell function.

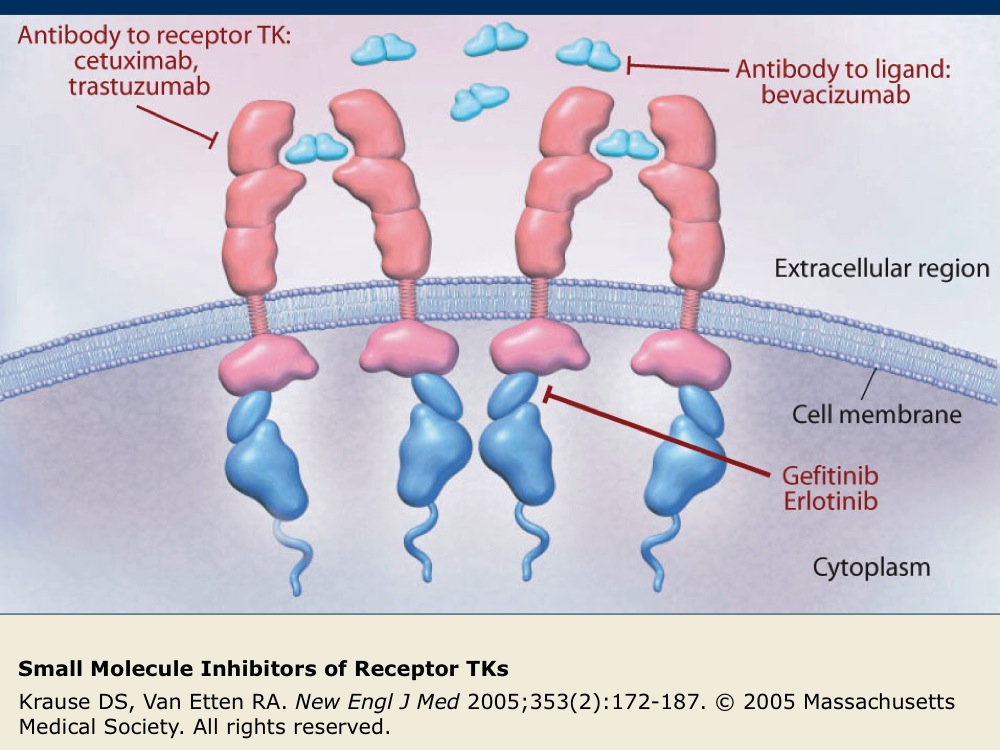
Small Molecule Inhibitors of Receptor TKs
Slide 6 of 10
Several small molecules that inhibit the function of receptor TKs (indicated in red) have been developed. Gefitinib and erlotinib are intracellular inhibitors of EGFR that block EGFR phosphorylation and subsequent activation. Cetuximab and trastuzumab are antibodies targeted against receptor TKs. Bevacizumab is an antibody directed against the VEGF-A ligand that prevents the interaction of VEGF-A with its receptor, VEGFR.
Inhibition of receptor TK function blocks the downstream signaling cascades of cellular processes, such as angiogenesis and cell proliferation, that can promote the survival of tumor cells.
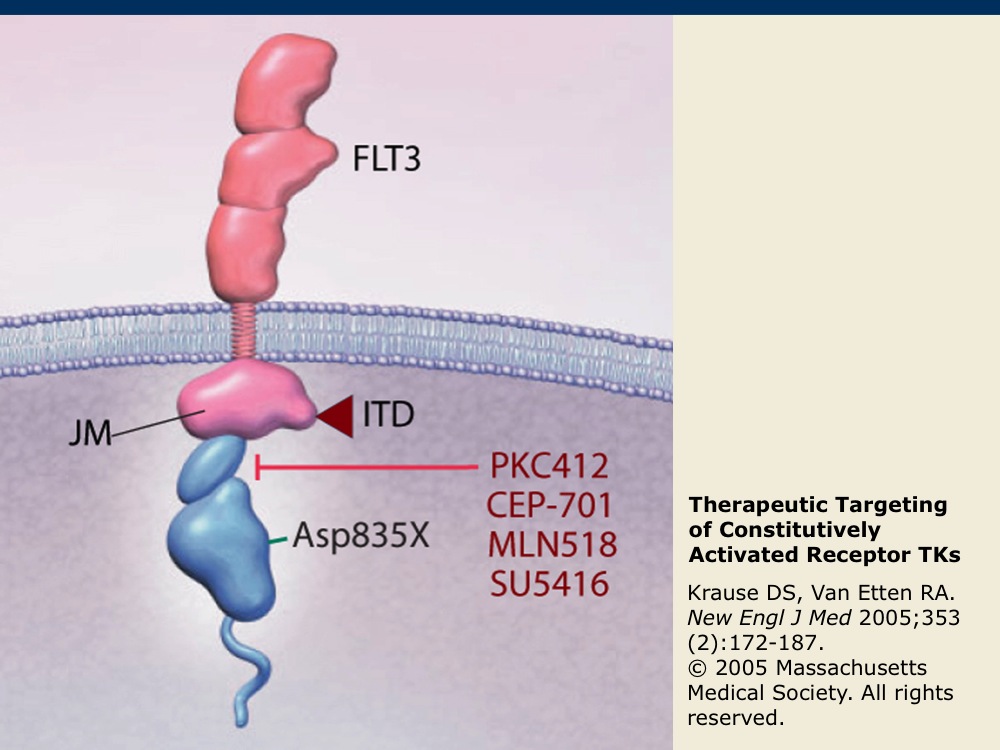
Therapeutic Targeting of Constitutively Activated Receptor TKs
Slide 7 of 10
FLT3 is a receptor TK that is expressed on blast cells in the majority of acute myeloid leukemia (AML) cases. It may become constitutively activated by internal tandem duplications (ITD) or by a point mutation (D835X) within the receptor TK. FLT3 is also among the receptor TKs that are targeted by sorafenib and sunitinib. Inhibitors of FLT3 kinase activity under development, such as midostaurin (PKC412), lestaurtinib (CEP-701), tandutinib (MLN518) and semaxanib (SU5416), are indicated in red.
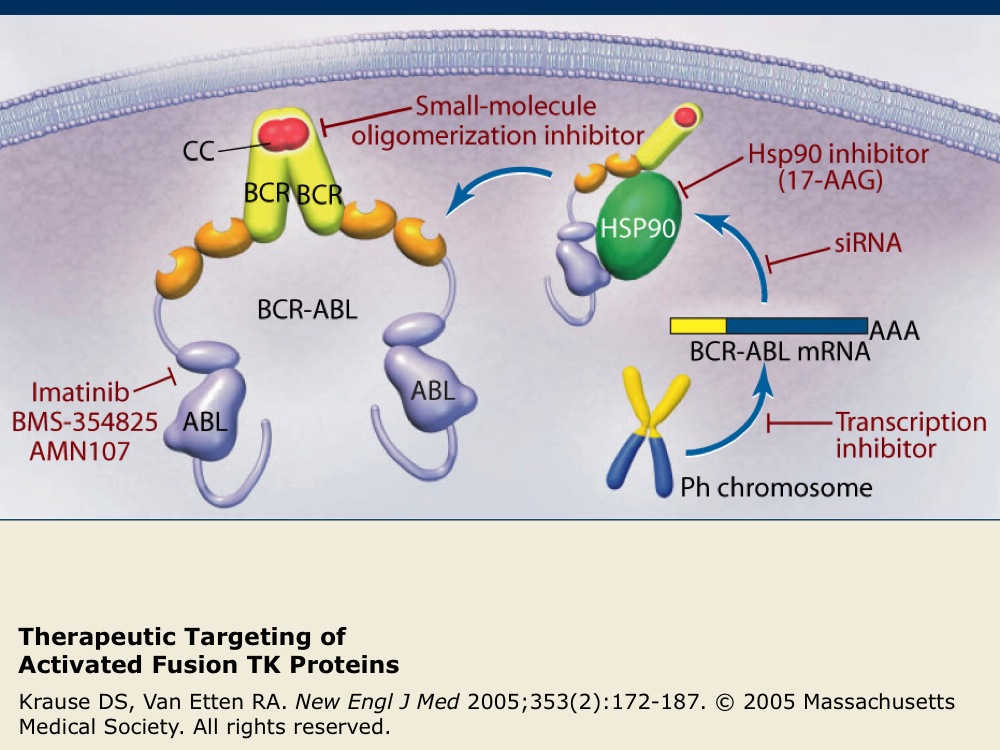
Therapeutic Targeting of Activated Fusion TK Proteins
Slide 8 of 10
The activated TK fusion protein BCR-ABL is the only proven molecular target for chronic myeloid leukemia (CML). Examples of therapeutic agents targeting the activated TK fusion protein BCR-ABL are listed in red.
Imatinib is a specific inhibitor of several TKs, including ABL, c-KIT and PDGFR, that has been successful as a treatment for CML. Nilotinib is also a highly selective inhibitor of BCR-ABL kinase activity that has been recently shown to have superior efficacy compared to imatinib for the treatment of patients with CML in the chronic phase.
17-AAG interferes with binding to cellular chaperone proteins such as Hsp90. Small interfering RNAs (siRNA) act to promote the degradation of BCR-ABL mRNA and other agents act to block oligomerization or BCR-ABL transcription.

Limitations of TK-Targeted Therapies: Development of Resistance
Slide 9 of 10
Though advances in the development of TK-targeted therapies have been made, resistance to these therapies is a growing problem. Causes of imatinib resistance in chronic myeloid leukemia are depicted, but the mechanisms involved in the development of resistance are applicable to both small-molecule TK inhibitors and to monoclonal antibodies directed against receptor TKs.
(A) There may be increased efflux of the drug from the cancer cell mediated by membrane transporter proteins that result in a decreased intracellular concentration of the drug.
(B) Proteins in the blood plasma may bind the drug and decrease its effective concentration.
(C) The TK molecule targeted by the drug may gain a mutation that allows for it to escape the drug’s action.
(D) Mutations may result in the activation of components of the signaling pathway downstream of the targeted TK.
(E) Amplification of the TK gene resulting in overproduction of the TK can confer relative resistance to an inhibitor.

Mechanism of Action of Inhibitors of the VEGF/VEGFR Signaling Pathway
Slide 10 of 10
In addition to bevacizumab, which targets the VEGF ligand, the VEGF/VEGFR signaling pathway can be inhibited by the small molecules sunitinib and sorafenib. These oral multikinase inhibitors interfere with the activation of VEGFRs 1 to 3 by preventing phosphorylation. Sorafenib also inhibits the activity of Raf-1 kinase that functions in the signaling pathway, which is initiated after VEGFR binds its ligand.
- «
- 01 01 Editor's Note
- 02 02
- 03 03 Regulation of TK Activity-Inactive State
- 04 04 Regulation of TK Activity-Activated State
- 05 05
- 06 06 Small Molecule Inhibitors of Receptor TKs
- 07 07 Therapeutic Targeting of Constitutively Activated Receptor TKs
- 08 08 Therapeutic Targeting of Activated Fusion TK Proteins
- 09 09 Limitations of TK-Targeted Therapies: Development of Resistance
- 10 10 Mechanism of Action of Inhibitors of the VEGF/VEGFR Signaling Pathway
- »
- Pause